Protein-related items¶
You can also look at the changing of secondary structure elements
during your run. For this, you can use the program
gmx do_dssp, which is an interface for the
commercial program DSSP 176. For
further information, see the DSSP manual. A typical
output plot of gmx do_dssp is given in
Fig. 60.

Fig. 60 Analysis of the secondary structure elements of a peptide in time.
One other important analysis of proteins is the so-called Ramachandran plot. This is the projection of the structure on the two dihedral angles \(\phi\) and \(\psi\) of the protein backbone, see Fig. 61:
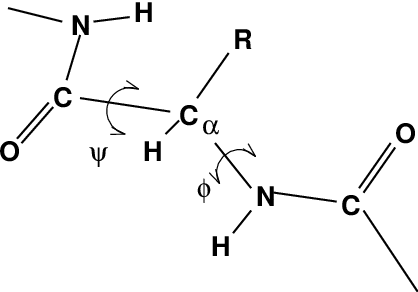
Fig. 61 Definition of the dihedral angles \(\phi\) and \(\psi\) of the protein backbone.
To evaluate this Ramachandran plot you can use the program gmx rama. A typical output is given in Fig. 62.
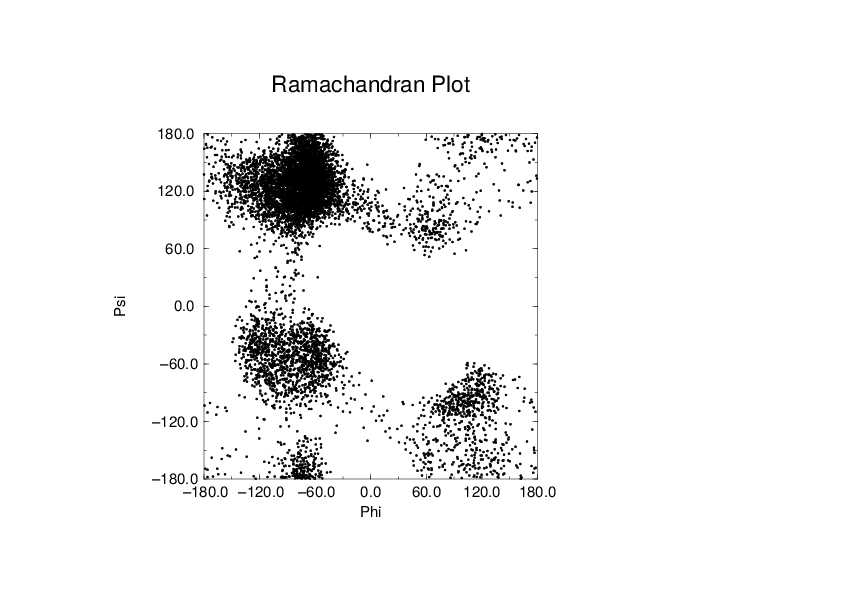
Fig. 62 Ramachandran plot of a small protein.
When studying \(\alpha\)-helices it is useful to have a helical wheel projection of your peptide, to see whether a peptide is amphipathic. This can be done using the gmx wheel program. Two examples are plotted in Fig. 63.
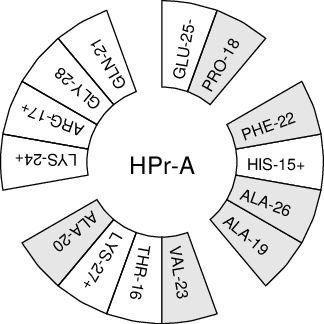
Fig. 63 Helical wheel projection of the N-terminal helix of HPr.