Protein-related items¶
To analyze the secondary structure of a protein (not only for static
structures, but also for trajectories), you can use the program
gmx dssp, which is a native implementation of DSSP
algorithm 176, but also is based on the (DSSP
V.4 algorithm) with some additional
features. For example, you can take into account native hydrogens from
the structure (-hmode gromacs, set by default), while in the original
algorithm, hydrogen atoms are set as pseudo-atoms with coordinates based
on the coordinates of the MainChain atoms (-hmode dssp). Also, it is
possible to conduct a fast search for neighboring residues using
Neighbor Search (-nb, default), instead of the slow enumeration of
protein residues among themselves according to the “each with each”
principle implemented in the original algorithm (-nonb).
One other important analysis of proteins is the so-called Ramachandran plot. This is the projection of the structure on the two dihedral angles \(\phi\) and \(\psi\) of the protein backbone, see Fig. 60:
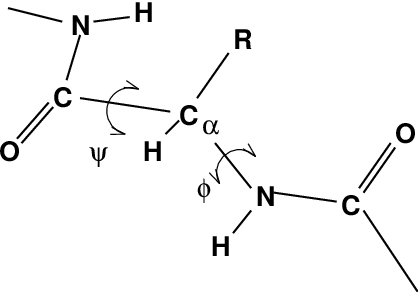
Fig. 60 Definition of the dihedral angles \(\phi\) and \(\psi\) of the protein backbone.¶
To evaluate this Ramachandran plot you can use the program gmx rama. A typical output is given in Fig. 61.
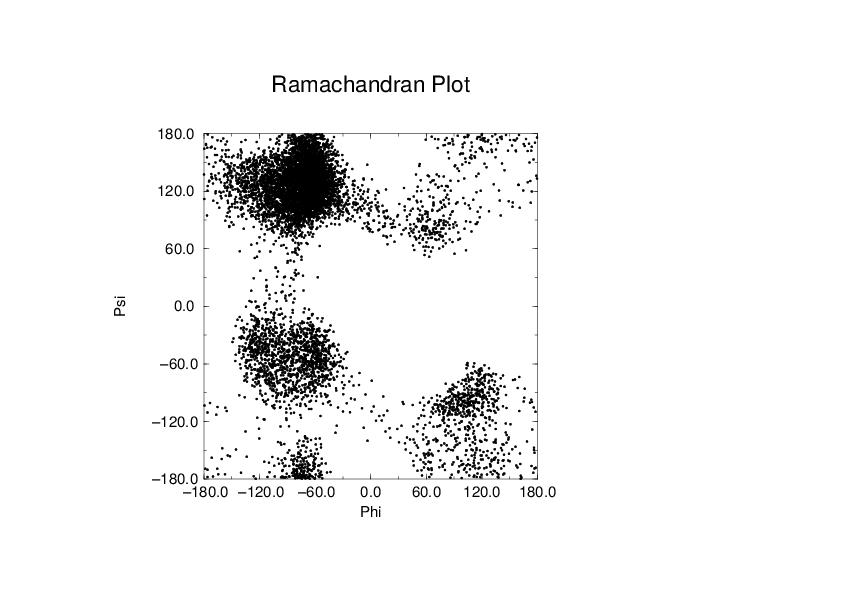
Fig. 61 Ramachandran plot of a small protein.¶
When studying \(\alpha\)-helices it is useful to have a helical wheel projection of your peptide, to see whether a peptide is amphipathic. This can be done using the gmx wheel program. Two examples are plotted in Fig. 62.
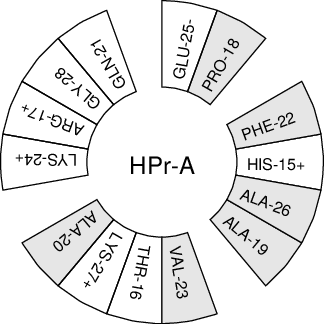
Fig. 62 Helical wheel projection of the N-terminal helix of HPr.¶